Demo
Run the demo with these simple steps » Get help on the Discord channel
Download the necessary files for demo
magnolia download demo --folder=demo
Parameters
--folder= relative path to where files are downloaded

Fig.1 - Alignment map images
Load the bam file into a set of training images
sudo magnolia load
--bam=NA12878-platinum-chr20.bam
--vcf=chr20_AF15_3.vcf
--ref=chr20.fa
--region=chr20:70000-6000000
--label=AF --folder=demo
--verbose --display
Result
This generated three image sets of about 3K samples each:
ls demo
ls demo/AF=0.5 | wc
ls demo/AF=1 | wc
ls demo/none | wc
Train deep model on these labels
sudo magnolia train --folder=demo --verbose --display
Result
After 1000 iterations this results in a model file:
ls -lh demo/model.pb
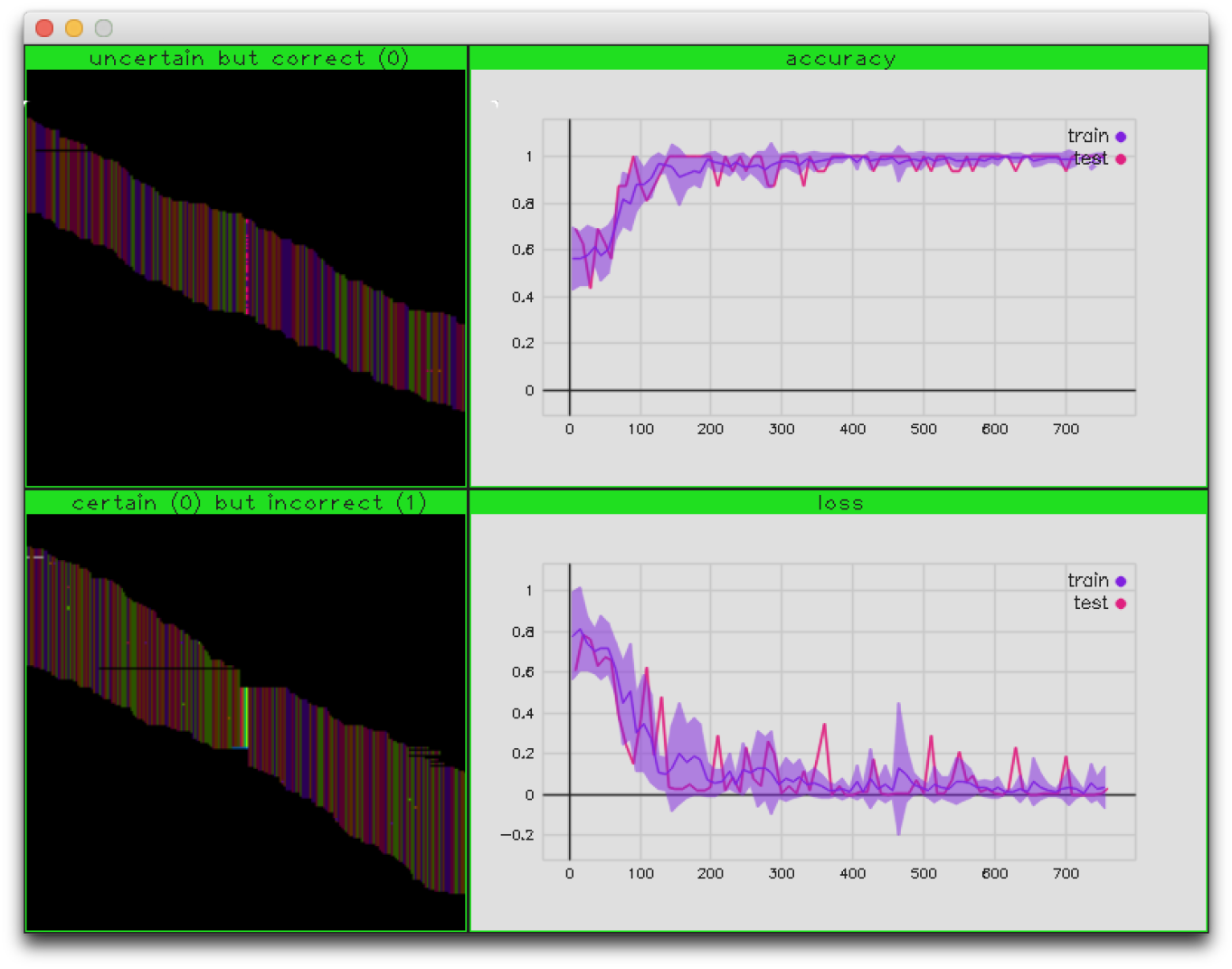
Fig.2 - Variant training display
Call a bam file not seen before.
To generate a .vcf based on the model: sudo magnolia call
--bam=NA12878-platinum-chr20.bam
--ref=chr20.fa
--region=chr20:70000-6000000
--label=AF
--folder=demo
--output=output.vcf
--verbose --display
Result
This outputs:
ls -lh demo/output.vcf
head -n20 demo/output.vcf



